2018-1-3 19:00 |
Вторая часть рассказа о самых интересных научных событиях 2017 года посвящена открытиям, связанным со здоровьем человека. Победы генной медицины 2017 год принес целый ряд значимых успехов в области генной терапии различных заболеваний, в том числе и таких, что считаются неизлечимыми.
В феврале китайские ученые сообщили об успешном эксперименте по применению технологии редактирования генома CRISPR-Cas9 для генетического заболевания кожи - дистрофического буллезного эпидермолиза (ДБЭ), при котором кожа становится очень ранимой, на ней часто возникают наполненные жидкостью пузыри и зоны эрозии, превращающиеся в обширные раны. Существует более десятка вариантов мутаций, ведущих к буллезному эпидермолизу. Они встречаются с разной частотой от одного на 50 тысяч до одного на миллион новорожденных. Некоторые формы болезни могут быть летальными в младенчестве, а другие увеличивают вероятность агрессивного рака кожи. Единственный способ лечения - регулярная обработка ран. В США расходы больного на перевязочные материалы составляют до 100 000 долларов в год. Также им регулярно нужны средства для обработки ран, антибиотики, обезболивающие препараты и другие лекарства. Больных ДБЭ часто называют «дети-бабочки», поскольку их кожа столь же хрупка и ранима, как крыло бабочки. В эксперименте китайских ученых были использованы лабораторные мыши, у которых присутствовала мутация в аналогичном гене, вызывающая схожие клинические проявления. По замыслу исследователей, нужно было вырезать из генома участок, содержащий мутацию, в результате чего должен был получиться работающий вариант гена, который обеспечил бы синтез пусть несколько измененного, но способного выполнять свои функции белка. В клетки эпидермиса новорожденных мышей был введен комплекс из белка Cas9, который способен вырезать участок генома, и РНК гида, направляющего белок Cas9 к нужному участку ДНК. Когда это было сделано, связь между слоями кожного эпителия у подопытных мышей значительно возросла. Авторы работы полагают, что усовершенствованную ими методику редактирования генома можно будет применить для лечения других наследственных заболеваний в различных тканях организма. К тому же в данном варианте метода значительно снижена вероятность того, что белок Cas9 повредит другие гены.
Другое сообщение об использовании генной терапии для лечения «детей-бабочек» поступило в конце года от ученых Германии и Италии. Семилетний мальчик, страдающий редким генетическим заболеванием, получил новую кожу благодаря генетически модифицированным стволовым клеткам. Его состояние остается хорошим спустя уже два года после лечения. Новый подход к лечению эпидермолиза аналогичен уже существующему методу лечения тяжелых ожогов, когда слои здоровой кожи выращиваются из собственных клеток пациента и пересаживаются на тело. Но команда под руководством Мишеля де Луки из Университета Модены и Реджо-Эмилии снабдила клетки, используемые для получения трансплантантов, новыми генами, которые свободны от мутаций, вызывающих буллезный эпидермолиз. Первая попытка лечения болезни была предпринята ими в 2006 году, когда они успешно помогли пациенту с небольшими поражениями кожи на ногах. Об этом они тогда сообщили в журнале Nature. Затем они сумели помочь еще одному пациенту
В 2015 году к Мишелю де Луке и его команде обратились коллеги-врачи из Германии, у которых был пациент с тяжелой формой буллезного эпидермолиза, при которой поражается светлая пластинка базальной мембраны (lamina lucida) - тонкий слой между соединительной тканью и кожным эпителием. Вызывается эта разновидность болезни мутацией в гене LAMB3 в первой хромосоме, кодирующем часть белка ламинина. Состояние мальчика было тяжелым. Из-за постоянно слезающей кожи он заразился несколькими инфекциями.
Мишель де Лука выделил эпидермальные клетки из неповрежденного участка кожи размером не более почтовой марки в паху мальчика. В культуре клеток имелись и клетки-предшественницы - стволовые клетки, “специализирующиеся” на определенном типе ткани, в данном случае на коже. Благодаря им в организме происходит постоянное обновление кожи. В лаборатории де Лука инфицировал эти клетки ретровирусом, который нес нормальную копию гена ланимина. Затем из этих клеток выращивали целые куски кожи размером от 50 до 150 квадратных сантиметров. Врачи из Рурского университета в городе Бохум провели две операции, пересаживая эти куски новой кожи на руки, спину, грудь и ноги мальчика. Через месяц после операций новая кожа начала восстанавливаться, покрыв 80 % тела мальчика крепким и эластичным эпидермисом. На пересаженных участках в течение двух прошедших с тех пор лет не возникают новые пузыри. Мальчик хорошо себя чувствует и даже играет в футбол.
В марте во Франции генная терапия была впервые в истории успешно применена для лечения серповидноклеточной анемии. В мире ежегодно рождается около 300 тысяч детей с этой болезнью. Она вызывается мутацией в гене, кодирующем одну из субчастиц молекулы гемоглобина. Она приводит к изменению формы белковой молекулы в красных клетках крови (вместо гемоглобина A появляется гемоглобин S). Эритроциты с дефектным гемоглобином имеют характерную серповидную форму, тогда как нормальные эритроциты имеют форму диска с вогнутой сердцевиной.
Серповидные эритроциты чаще обычных разрушаются, что влечет постоянную анемию, так как из-за гибели эритроцитов ткани недополучают кислород. К тому же такие эритроциты менее пластичны и хуже способны проходить через узкие капилляры. Частая закупорка капилляров у больных приводит к различным нарушениям, появлению тромбоза в печени и селезенке, артриту, язвам на ногах к тому же для них всегда характерна анемия и постоянная усталость. В некоторых случаях закупорка капилляров сетчатки глаза может привести к слепоте. Единственной формой непаллиативного лечения для таких больных до последнего времени была пересадка донорского костного мозга, но не для всех из них удается найти подходящего донора, к тому же трансплантация требует пожизненного применения препаратов, снижающих иммунитет.
Новый метод был сначала успешно испытан на животных, а в 2014 году в Детской больнице Неккер в Париже было начато первое клиническое испытание на человеке. Ученые работали с тринадцатилетним мальчиком с тяжелой формой серповидноклеточной анемии. Из-за последствий болезни у него развился двусторонний остеонекроз тазобедренного сустава, он также перенес удаление селезенки и желчного пузыря. Исследователи взяли у мальчика образец ткани его костного мозга. Из ткани были выделены мультипотентные гематопоэтические клетки. При помощи специально сконструированного вируса они внесли в его клетки гены другой формы гемоглобина. Причем они использовали не просто ген гемоглобина A, а добились замены в молекуле этого белка одной аминокислоты (глутамин на треонин). Этот вариант гена был обнаружен у некоторых детей, у которых ожидалось появление серповидноклеточной анемии, но ее симптомы не проявлялись. После этого ученые использовали препарат бусульфан, чтобы уничтожить оставшиеся в организме мальчика кроветворные клетки. Такую процедуру обычно делают перед пересадкой костного мозга. Затем мальчику вернули взятые у него клетки с модифицированным теперь геном, ответственным за гемоглобин.
Примерно через полгода после процедуры уровень гемоглобина в крови мальчика достиг 106 - 120 граммов на литр (при норме для его возраста 110 - 160 граммов на литр). Частично это был гемоглобин S, частично - нормальный гемоглобин A. Через девять месяцев нормальный гемоглобин составлял 46 % от всего гемоглобина в организме, а спустя 15 месяцев после пересадки клеток нормального гемоглобина было уже 48 %. Такого количества гемоглобина А достаточно, чтобы симптомы серповидноклеточной анемии исчезли. На протяжении всех 15 месяцев у пациента не наблюдалось проявлений болезни. И врачи смогли отменить дополнительную терапию лекарствами и переливания крови. Исследователи будут в дальнейшем лишь наблюдать за состоянием крови мальчика, чтобы узнать, сохранится ли нормальный вариант гемоглобина в его организме и далее. Пока же можно сказать, что мальчик, в течение многих лет страдавший от тяжелой болезни, избавился от нее.
В сентябре Управление по контролю качества пищевых продуктов и лекарственных препаратов США (FDA) одобрило применение генной терапии для лечения острого лимфобластного лейкоза. Это первый случай, когда метод, связанный с модификацией генома, войдет в клиническую практику США. Терапевтический метод, получивший коммерческое название Kymriah (ранее известен как tisagenlecleucel), разработан крупной фармацевтической компанией Novartis. Он основан на применении Т-лимфоцитов с химерными антигенными рецепторами (chimaeric antigen receptor, CAR). Общее название подобных методов лечения - CAR T-терапия.
У больного лейкозом берутся его собственные Т-лимфоциты, они замораживаются и доставляются в исследовательский центр Novartis в Нью-Джерси. Там, в лаборатории, при помощи специально созданного вируса Т-клеткам передается ген определенного белка - поверхностного рецептора клетки. Данный белок способен избирательно связываться с антигенами - белками раковых клеток. Таким образом, Т-лимфоциты, вернувшись в организм пациента, уже обучены распознавать и уничтожать раковые клетки. В случае острого лимфобластного лейкоза такими клетками становятся безудержно размножающиеся B-лимфоциты, а мишенью для химерного антигенного рецептора - их поверхностный белок CD19. Фактически, врачи, возвращая генетически модифицированные T-клетки в организм пациента, «обучают» его собственную иммунную систему бороться с лейкозом. От забора Т-клеток у пациента до их возвращения будет проходить 22 дня.
Модифицированные Т-клетки уничтожают все B-клетки, поэтому пациент, к сожалению, не только избавится от лейкоза, но и утратит способность вырабатывать антитела к другим антигенам (за эту способность во многом отвечают именно B-клетки). Поэтому ему затем придется каждые несколько месяцев получать инъекции донорских антител для поддержки своего иммунитета. Это хлопотно, но лучше, чем тяжелая форма лейкемии.
Первое исследование химерных антигенных рецепторов было проведено 26 лет назад - в 1991 году - учеными из Калифорнийского университета в Сан-Франциско. Тогда их использование было недостаточно эффективным для борьбы с лейкозом. Но за прошедшее время метод многократно улучшили. Исследования показывают, что терапия с использованием CAR Т-клеток может давать длительные ремиссии в тех случаях, когда другие способы лечения оказываются бессильны. В клиническом испытании, начавшемся в 2015 году, ремиссия в течение трех месяцев наблюдалась у 82,5% из 63 пациентов. Через полгода доля выживших составляла 89 %, через год - 79 %. Напомним, что модифицированные лимфоциты применялись только в тех случаях, когда химиотерапия не работала или после нее возникали рецидивы.
Kymriah планируется применять при B-лимфоцитном остром лейкозе у пациентов младше 25 лет, при лечении которых оказалась неэффективной химиотерапия. Отметим, что на долю B-лимфоцитов приходится более 80 % всех случаев острого лимфобластного лейкоза, а наибольшее число случаев этого заболевания приходится на детский возраст. Согласно оценкам Национального института рака США, в стране ежегодно появляется около 3100 больных острым лимфобластным лейкозом в возрасте 20 лет и младше, примерно у 600 заболевание дает рецидив после химиотерапии и во многих случаях остается неизлечимым.
Правда, стоимость лечения Kymriah составляет 475 тысяч долларов только за саму процедуру по генетической модификации клеток, без учета оплаты работы врачей и других расходов. Novartis создал фонд для пациентов, медицинская страховка которых не оплачивает такое лечение, а также обещает не брать плату, если терапия для данного пациента не окажется эффективной по прошествии месяца. Novartis вряд ли рассчитывает получить от нового метода сверхприбыли. Слишком мало число пациентов в год, для которых он будет применяться. Скорее компания заинтересована в первом официально разрешении генной терапии в США как таковом, что в дальнейшем должно проложить дорогу генным методам лечения других болезней. Чтобы упрочить позиции генной терапии в глазах регулирующих органов (а мнение FDA, пусть неофициально, будут учитывать и аналогичные организации в странах Европы), нужны статистические данные о долговременной безопасности этого метода.
Другим подводным камнем на пути CAR T-терапии явлется острая воспалительная реакция, известная как «цитокиновый шторм». При активизации Т-лимфоцитов ими выделяются цитокины - сигнальные вещества, которые активируют другие клетки иммунной системы, способные убивать чужеродные клетки. При этом у больного поднимается температура, возникает лихорадка, может резко упасть давление и даже развиться отек легких.
Представители Novartis, однако, утверждают, что опытные врачи в состоянии справиться с этой проблемой. Также у некоторых больных в ходе лечения наблюдались неврологические расстройства: судорожные припадки и галлюцинации, но они носили временный характер. На данный момент FDA разрешило применять метод Kymriah для пациентов только в тех больницах, где есть достаточно квалифицированный персонал, чтобы оказать своевременную помощь при «цитокиновом шторме». В течение месяца Novartis намерен сделать свой метод доступным в 20 больницах на территории США, позднее их число планируют увеличить до 32.
В октябре Консультативный комитет FDA рекомендовал одобрить еще один метод генетической терапии - лечение амавроза Лебера, врожденной патологии сетчатки, которая неминуемо ведет к полной слепоте. У людей с амаврозом Лебера и органы зрения, и нервные пути, ведущие от глаза в мозг, и отвечающие за зрения нейроны мозга в полном порядке. Однако зрение у них утрачивается, поскольку в их сетчатке гибнут и не восстанавливаются светочувствительные клетки. Причина этого кроется в мутации гена, ответственного за правильное развитие этих клеток, чаще всего это ген RPE65. Существует 189 различных вариантов мутаций, ведущих к амаврозу Лебера. Правда, 146 из них встретились на практике всего по одному разу, а за 70 % случаев болезни отвечают всего восемь типов мутаций. В зависимости от конкретной мутации человек может уже родиться слепым, утратить зрение в первые месяцы жизни, а есть и случаи, когда клетки сетчатки гибнут медленно, и тогда слепота наступает примерно к 30 годам. До появления генной терапии амавроз Лебера был полностью неизлечим. Опыты по лечению амавроза Лебера начались во середине 2000-х годов. Сначала они проводились на животных, у которых имелись аналогичные мутации, нарушающие восстановление клеток сетчатки. В 2006 году Сью Семпл-Роуланд из Флоридского университета и ее коллеги смогли восстановить зрение шести из семи подопытных цыплят. Нужная генетическая конструкция вводилась им в эмбриональном периоде при помощи специально сконструированного вируса. Французские ученые в том же году провели успешный эксперимент на собаках.
В 2008 году о своем эксперименте по применению генной терапии амавроза Лебера у людей рассказали сотрудники Офтальмологической больницы Мурфилдса и Института офтальмологии Университетского колледжа Лондона. Коллектив ученых работал с пациентами, у которых была разновидность амавроза Лебера, проявляющаяся не сразу после рождения, а постепенно. У них было три пациента возрастом от 17 до 23 лет с мутацией в гене RPE65 и нарастающей потерей зрения. Имя сделали инъекцию вируса, содержащего исправленную версию гена, под сетчатку глаза (так называемая «субретинальная инъекция»). Спустя несколько месяцев проверка показала, что у двух пациентов улучшения зрения не последовало, но и прогресса болезни не наблюдалось. А вот у третьего, 17-летнего Стивена Ховарта, зрение значительно улучшилось. До терапии Стивен испытывал особые трудности с сумеречным зрением. Ночью в городе он мог видеть только яркие пятна света: уличные фонари и фары автомобилей. В слабоосвещенной комнате он двигался только ощупью, а на улицу по ночам и вовсе не выходил. После генной терапии он стал с легкостью преодолевать специальный затемненный лабиринт, в котором раньше проходил лишь метр за минуту. На ночной городской улице Стивен стал различать тротуар, дорожную разметку, стены домов, дорожные знаки, даже трещины в асфальте. Он осмелел настолько, что решился совершать ночные прогулки по родному городу.
Для генной терапии амавроза Лебера используется модифицированный аденоассоциированный вирус, который выращивается в клеточной культуре HEK 293. Вирус снабжается комплиментарной ДНК, содержащий работающую версию гена RPE65. В 2009 году были опубликованы результаты первой фазы клинических испытаний этого метода терапии. Участвовало 12 пациентов возрастом от 8 до 44 лет. В качестве меры предосторожности вирусный препарат вводился только в один из глаз. Улучшение зрения в итоге наблюдалось у всех двенадцати пациентов, причем у детей прогресс был большим, чем у взрослых.
После успешных результатов последующих опытов, в 2013 году в Пенсильванском университете была создана компания Spark therapeutics для доведения этого метода терапии до реального применения. Метод назвали Voretigene neparvovec (сейчас в качестве коммерческого названия взято слово Luxturna). Параллельно во Флориде Сэмюэль Джекобсон и Уильям Хаусвирт провели независимое испытание на двенадцати пациентах при поддержке Национального института зрения США. В отличие от пенсильванских экспериментов инъекция препарата делалась не в центральную ямку сетчатки, которая в норме является ее самым чувствительным к свету участком и обеспечивает центральное поле зрение (желтое пятно), а чуть ниже. В результате у пациентов зрение улучшалось, но область самого четкого зрения оказывалась смещенной к тому месту, где был введен препарат.
Недавно Spark therapeutics опубликовала результаты уже третьей фазы клинических испытаний. Они вновь оказались успешными. По расчетам ученых модифицированный вирус может сохраняться в зоне сетчатки и обеспечивать правильную работу гена длительное время, в течение как минимум нескольких лет. Теперь заявку компании на официальную регистрацию своего метода рассмотрел состоящий из шестнадцати человек консультативный комитет по клеточной, тканевой и генной терапии при FDA. Это независимый орган и его решения служат только рекомендацией для работников FDA, но вес этих рекомендаций очень высок. За одобрение генной терапии амавроза Лебера члены комитета высказались единогласно. Теперь до 12 января 2018 года FDA должно принять официальное решение, одобрить или нет применение этого метода в клинической практике в США. Если ответ будет положительным, то Luxturna станет первым разрешенным в США методом генной терапии, в котором используется вирусный носитель.
Следует помнить, что Luxturna не излечивает амавроз Лебера полностью и не восстанавливает утраченные клетки сетчатки. Зрение пациентов ограничивается возможностями тех жизнеспособных клеток, которые сохранились у них на момент инъекции. Именно поэтому у детей терапия дает большее улучшение, чем у взрослых. Не до конца ясно, насколько будет долгим эффект.
Тоже в октябре 2017 года FDA разрешило применение на территории страны еще одного вида генной терапии. Он предназначен для борьбы с B-клеточной лимфомой. Он получил коммерческое название Yescarta (ранее известен как аксикабтаген цилолейкел, axicabtagene ciloleucel). Разработан метод компанией Kite Pharma, которую в августе этого года приобрела более крупная биофармацевтическая корпорация Gilead Sciences. Применяться Yescarta будет при лечении больных, для которых оказались неэффективными по крайней мере два других метода терапии, например, химический препарат и облучение. Новый метод будет применяться в случаях диффузной B-крупноклеточной лимфомы (diffuse large B-cell lymphoma, DLBCL). Это самый распространенный тип неходжкинской лимфомы у взрослых пациентов, в США и Великобритании в год на 100 тысяч человек насчитывается 7 - 8 случаев этой болезни. DLBCL - это агрессивная опухоль, которая может возникать практически в любой части тела и отличается быстрым ростом.
Как и Kymriah, метод Yescarta основан на использовании химерных антигенных рецепторов. Для каждого пациента препарат нужно готовить индивидуально. У больных забирают их собственные лимфоциты и в лаборатории при помощи специально сконструированного вируса наделяют их геномом белка - поверхностного рецептора клетки, который способен избирательно связываться с белками опухолевых клеток. В данном случае мишень служит белок CD19 на поверхности B-лимфоцитов. Затем эти клетки возвращают в организм пациента, где они начинают сами бороться с лимфомой. В клинических испытания у 72 % пациентов после применения Yescarta количество опухолевых клеток снизилось, а у 51 % дело дошло до полной ремиссии.
Еще одной октябрьской новостью стало сообщение Национальной службы здравоохранения Великобритании, что она будет оплачивать генную терапию врожденного иммунодефицита. Метод Strimvelis, разработанный компанией GlaxoSmithKline, станет первым видом генной терапии, получившим государственное финансирование в Великобритании. Хотя цена такого лечения составляет более 500 тысяч фунтов стерлингов специалисты признали его более эффективным и, в конечном счете, более финансово оправданным по сравнению с терапией при помощи регулярного приема лекарств.
Метод лечения предназначен для борьбы с дефицитом аденозиндезаминазы (ADA deficiency, ADA-SCID). Он встречается примерно у одного новорожденного на сто тысяч. Из-за мутации в двадцатой хромосоме у больных в организме не вырабатывается фермент аденозиндезаминаза, участвующий в синтезе пуринов. По этой причине нарушается работа естественного иммунитета, и человек становится уязвимым для любой инфекции. Лишь половина детей с этим генетическим расстройством доживает до шести месяцев.
Сразу после рождения такого ребенка необходимо поместить в пластиковый кювет, внутри которого создается стерильная обстановка. В дальнейшем дети вынуждены расти только в защищенных помещениях, а, выходя наружу, носить специальные защитные костюмы, поэтому эту болезнь иногда называют «синдром ребенка в пузыре». Частично облегчить состояние больных может ферментная заместительная терапия - постоянный прием препаратов, заменяющих недостающий фермент. В некоторых случаях помочь больным может пересадка костного мозга.
Первое в истории успешное применение генной терапии было связано именно с лечением АДА-дефицита. Это произошло еще в 1990 году, когда в Бетесде (Мэриленд, США) Уильям Андерсон при помощи модифицированного вируса снабдил исправной копией гена четырехлетнюю девочку Ашанти Де Сильву. Лечение было успешным, по сообщениям 2007 года Ашанти Де Сильва была по-прежнему здорова, хотя и при условии регулярного приема лекарств.
В 2000-е годы последовал еще ряд успешных случаев применение генетической терапии при АДА-дефиците, а также при некоторых других видах врожденного иммунодефицита. Ученые из миланской клиники Сан-Рафаэле, а также компаний GlaxoSmithKline и MolMed создали метод Strimvelis, который годился для широкой клинической практики. Работа над ним велась с 2002 года. 75% детей, участвовавших в клинических испытаниях Strimvelis, не нуждались в дальнейшей ферментной заместительной терапии.
Метод состоит в получении у пациента гемопоэтических стволовых клеток, из которых для дальнейшего использования берутся только клетки, экспрессирующие мембранный белок CD34. Эти клетки культивируют с цитокинами и факторами роста, а затем при помощи вируса вводят в них ген аденозиндезаминазы. Затем клетки возвращают в организм пациента. Они укореняются в костном мозге, делятся и создают зрелые клетки с нормальным геном аденозиндеаминазы. Пока производить все требуемые манипуляции могут только в миланской больнице Сан-Рафаэле.
В подобном лечении нуждаются ежегодно 14 - 15 человек в странах Евросоюза и 12 человек в США. Цена курса высокая - 594 тысяч евро, но по сравнению с регулярными инъекциями ферментной заместительной терапии выходит дешевле, так как подобные инъекции в течение десяти лет жизни будут стоить не менее 4,25 миллиона долларов. Генная терапия проводится всего один раз и должна после этого действовать всю жизнь пациента. В апреле 2016 года комитет Европейского агентства по лекарственным средствам рекомендовал одобрить применение Strimvelis для детей с дефицитом аденозиндезаминазы, у которых нет подходящего донора костного мозга.
Теперь же в Великобритании лечение АДА-дефицита при помощи генной терапии будет оплачиваться Национальной службой здравоохранения. Национальный институт здоровья и клинического совершенствования опубликовал проект руководства, где указывается, что Strimvelis имеет лучшие показатели выживаемости пациентов по сравнению с заместительной терапией и обычной трансплантацией стволовых клеток. Авторы документа утверждают, что такое лечение даст детям больше шансов жить нормальной жизнью, ходить в школу и общаться с друзьями, не опасаясь опасной для жизни инфекции.
Наконец, в декабре 2017 года исследователи сообщили об первом в истории успешном эксперименте по излечению гемофилии при помощи генной терапии. В исследовании участвовало десять человек с гемофилией B. Это наследуемая болезнь, вызывается мутацией в гене, который заведует синтезом одного из белков, связанных со свертыванием крови, так называемого «фактора свертывания IX». В зарубежной литературе она иногда называется Christmas disease, так как первого пациента, у которого было описано данное заболевание звали Стивен Кристмас, а опубликовали описание в рождественском номере «Британского медицинского журнала». Гемофилия B - вторая по распространенность разновидность гемофилии, она встречается у 20 % гемофиликов и уступает только гемофилии A. Различие этих двух болезней было обнаружено лишь в 1952 году.
Именно гемофилия B была у потомков королевы Виктории - представителей правящих династий Германии, Испании и России, включая цесаревича Алексея Романова. Замена лишь одного нуклеотида в гене приводила к тому, что молекулы фактора свертывания IX получались неправильной формы и не могли нормально выполнять свои функции.
Участники нынешнего исследования получили однократную инъекцию безвредного вируса, который нес в себе нужный ген. При проверке через 18 месяцев оказалось, что уровень фактора свертывания IX в их печени вырос и составлял в среднем 34 % от нормы. Этого оказалось достаточно, чтобы у девяти из десяти пациентов не было кровотечений. Восемь из десяти больше не нуждались в ежедневных инъекциях фактора IX.
Предыдущие попытки применить генную терапию для лечения гемофилии B оказались неудачными, так как модифицированные клетки разрушались клетками иммунной системы организма или же производили слишком мало фактора IX. В этот раз авторы работы использовали версию гена, которая производить очень мощную разновидность этого белка, так что они смогли снизить дозу вируса при его занесении в организм, а значит минимизировать иммунную реакцию. Двое пациентов отреагировали на вирус повышением уровня ферментов печени, но этот эффект исчез после того, как они получили стероиды.
Помощь при болезни Хантингтона
Первые результаты испытаний свидетельствуют, что экспериментальное лекарство способно замедлить развитие болезни Хантингтона, наносящей непоправимый ущерб мозгу. Препараты, используемые в данный момент, помогают лишь смягчить симптомы этой болезни, но не влияют на ее течение.
Болезнь Хантингтона (Гентингтона) возникает из-за мутации в одном гене, контролирующем важный для функционирования нервной системы белок. Данный ген содержит довольно много идущих подряд троек нуклеотидов CAG (цитозин, аденин, гуанин). Когда число таких троек превышает 36, у человека начинаются проявление болезни. Причем от числа избыточных повторов зависит тяжесть заболевания. Обычно болезнь Хантингтона диагностируется в возрасте старше 30 лет. У больных наблюдаются перепады настроения, гнев и депрессия, характерны случаи непроизвольных беспорядочных движений (гиперкинезы), постепенно они учащаются, нарушается координация движений, появляются расстройства речи и сна, затем возникают когнитивные расстройства, проблемы с памятью, психические нарушения. С момента появления первых симптомов продолжительность жизни больного составляет около 15 - 20 лет.
Новый препарат, получивший название Ionis-HTTRx, разработан калифорнийской биотехнологической компанией Ionis Pharmaceuticals. Он представляет собой синтетическую одиночную нить ДНК, перехватывающую молекулу РНК, которая должна доставить в рибосомы информацию с мутантного гена. Благодаря этому синтез дефектного белка снижается, а значит, задерживается развитие болезни. В мозг препарат попадает с помощью инъекции в спинномозговую жидкость.
В испытаниях препарата участвовали 46 мужчин и женщин с разными стадиями болезни, живущие в Великобритании, Германии и Канаде. Пациентам были проведены четыре спинальные инъекции в течение месяца с увеличением дозы препарата. После приема концентрация дефектного белка в спинномозговой жидкости снижалась пропорционально дозе. Хотя данное испытание было слишком маленьким и недостаточно продолжительным, чтобы показать, улучшаются ли клинические симптомы, но оно дало основание для организации более масштабных испытаний. Если они окажутся успешными, возможно, препарат можно будет применять для регулярных инъекций, предотвращая проявление симптомов болезни у носителей мутантного гена.
Жировая ткань общается с другими органами
Новое исследование позволило узнать, что жировая ткань в организме может взаимодействовать с другими органами при помощи небольших молекул, которые контролируют активность генов в других частях тела. Этот новый путь связи в организме показывает, что жир играет большую роль в регуляции обмена веществ, чем считалось ранее. Также открытие может дать новые варианты лечения таких заболеваний, как ожирение и диабет.
Клетки жировой ткани
Ранее ученым уже было известно, что адипоциты - клетки жировой ткани - выделяют ряд гормонов, например, резистин, адипонектин и лептин. Но теперь выявлены новые носители информации - молекулы микроРНК. В организме микроРНК участвует в регуляции работы генов при помощи механизма так называемой РНК-интерференции (подробнее об этом явлении можно прочитать в специальном очерке «РНК и иммунитет»). Между клетками молекулы микроРНК путешествуют в экзосомах, небольших образованиях из цитоплазмы, имеющих мембрану. Когда экзосома достигает своей цели, микроРНК попадает в нужную клетку и работа генов в этой клетке корректируется.
Специалист по диабету Томас Томоу из Гарвардской медицинской школы и его коллеги решили уточнить функции микроРНК в жировой ткани. Для этого они создали особую линию генетически модифицированных лабораторных мышей, которые были лишены необходимого фермента для выработки микроРНК. Как оказалось, у этих мышей было меньше жировой ткани и они не были способны перерабатывать глюкозу так же эффективно, как обычные мыши.
Пересаживая этим мышам жир от обычных мышей, ученые восстановили уровень микроРНК у них в крови. При этом пересадка бурого жира позволила также нормализовать расщепление глюкозы в их организме, а вот при пересадке белого жира этого не происходило. Напомним, что обычно в организме белый жир служит для запаса энергии, а бурый, названный так из-за обилия митохондрий в клетках, используется для терморегуляции (функциям жировой ткани также посвящен отдельный очерк «Белый и бурый»).
В предыдущем исследовании мышей, у которых была нарушена функция образования микроРНК в жировой ткани, выяснилось, что у них также были затронуты сердце, печень и другие органы, хотя генетическая модификация не должна была повлиять непосредственно на них. Поэтому у Томоу и его коллег возникла гипотеза, что жировая ткань оказывает влияние на органы при посредстве молекул микроРНК.
Для проверки этого они подготовили две группы мышей. У одной из них клетки бурой жировой ткани могли продуцировать человеческие молекулы микроРНК. У другой в клетках печени производились молекулы, которые служат мишенью для этой микроРНК, связанные с флуоресцентным белком, чтобы их можно было легко обнаружить. После того, как экзосомы с микроРНК мышей первой группы были введены второй группе, число флуоресцирующих молекул в их печени упало. Это означает, что жировая ткань, выпуская в кровь микроРНК, способна влиять на экспрессию генов в печени. Также было обнаружено влияние этой микроРНК на важный для обмена веществ ген Fgf21 (ген фактора роста фибробластов 21) в клетках печени.
Как заморозить сердечную ткань
Успешный эксперимент по замораживанию и последующему размораживанию образцов сердечной ткани был проведен группой ученых в США. Использование магнитных наночастиц позволило избежать повреждения клеток при разморозке. В дальнейшем этот метод может быть применен для длительного сохранения целых органов.
Основной проблемой в криоконсервации органов оказывается разрушение живых тканей при размораживании. Согревание идет неравномерно, поэтому в клетках возникают маленькие кристаллы льда. Оболочки клеток разрываются или в них появляются трещины. В результате донорские органы могут быть использованы только в течение нескольких часов, пока клетки не умирают из-за отсутствия кровоснабжения. Каждый год в мире 60 % донорских сердец и легких становятся непригодными для трансплантации из-за истечения срока хранения. Ученые решили устранить эту проблему, применив магнитные наночастицы, которые, возбуждаясь в магнитном поле, обеспечивают быстрый и равномерный прилив тепла. Один из авторов работы Кельвин Брокбанк из Tissue Testing Technologies говорит, что теперь ученые видят путь к клиническому применению метода и сохранению тканий и органов для пересадки пациентам.
В эксперименте, описанном в журнале Science Translational Medicine, ученые охладили клапаны сердца и кровеносные сосудов свиней, используя криопротекторный раствор, смешанный с наночастицами оксида железа, покрытыми кремнием. Образцы охлаждали в жидком азоте до -160 °C. При оттаивании образец помещался внутри электромагнитной катушки, генерировавшей переменное магнитное поле. Под действием этого поля частицы колебались внутри образца и быстро и равномерно согревали ткани со скоростью от 100 до 200 °С в минуту, от 10 до 100 раз быстрее, чем существовавших ранее методах.
При испытании их механических и биологических свойств ткани не показали каких-либо признаков повреждений, в отличие от контрольных образцов, который согревались медленно. Исследователи также смогли успешно удалить из образцов наночастицы оксида железа. Сейчас исследователи планируют проверить новую методику криоконсервации на почках кролика и образцов кожи, мышц и кровеносных сосудов человека.
Сотворение эмбриона
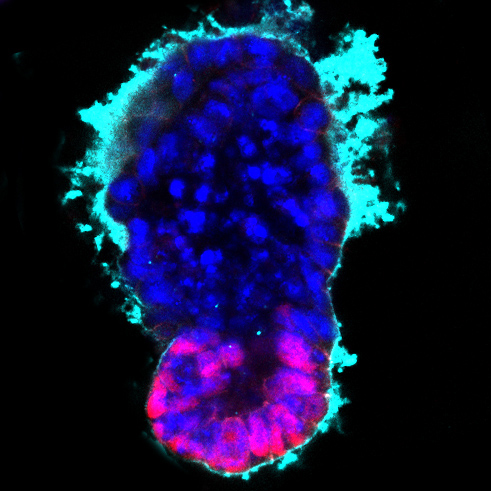
Ученые из Кембриджского университета сообщили о создании искусственного эмбриона мыши без использования половых клеток. Они смогли вырастить в лабораторных условиях структуру, напоминающую естественный эмбрион, применив только два типа стволовых клеток и внеклеточный матрикс.
При обычном развитии эмбриона млекопитающих, оплодотворенная яйцеклетка многократно делится, образуя эмбриональные стволовые клетки. Спустя некоторое время, когда наступает стадия бластоцисты, в составе эмбриона выделяются две группы клеток. Одна из них (эмбриобласт) образована клетками, которые в будущем образуют тело нового живого существа, другая (трофобласт) даст начало плаценте, с помощью которой эмбрион прикрепится к стенке матки и будет получать питание. На стадии бластоцисты эмбрион имеет вид полого шарика (у человека его диаметр равен примерно одной десятой миллиметра). Оболочку этого шарика образуют клетки трофобласта, а клетки эмбриобласта скапливаются внутри, возле одного из его полюсов.
В предыдущих попытках вырастить искусственный эмбрион, не прибегая к искусственному оплодотворению или клонированию, использовались только стволовые клетки эмбриобласта. В результате не удавалось получить структуру, хотя бы отчасти напоминающую эмбрион на ранней стадии развития, так как в этот период требуется, чтобы разные типы клеток координировали тесное взаимодействие друг с другом.
В новом исследовании авторы использовали не только стволовые клетки эмбриобласта, но и трофобластные стволовые клетки (их также называют экстраэмбриональными стволовыми клетками), а также внеклеточный матрикс, который должен был обеспечить трехмерную структуру эмбриона.
Внеклеточный матрикс - это каркас любого органа, окружающий каждую клетку. Обычно он состоит из коллагена и других гликопротеинов, протеогликанов и гиалуроновой кислоты. В последние несколько лет матрикс все чаще используется в экспериментах по созданию новых органов. Разработан способ вымывать все клетки из матрикса, оставляя сам каркас неповрежденным. После этого материкс заселяется стволовыми клетками, которые формируют орган. Это может найти применение в трансплантологии, когда донорский орган нельзя пересадить пациенту напрямую из-за иммунного отторжения, но можно взять матрикс, удалить из него клетки донора (сам по себе матрикс иммунного ответа не вызывает) и заполнить стволовыми клетками реципиента.
В данном случае матрикс был заселен трофобластными и эмбриобластными стволовыми клетками. В результате ученые смогли вырастить структуру способную к самостоятельному развитию, причем ее строение было весьма близко к естественному зародышу мыши. «И эмбриональные, и внеэмбриональные клетки начинают говорить друг с другом и становятся организованными в структуру, которая выглядит и ведет себя как эмбрион, - объясняет профессор Магдалена Зерницка, которая возглавляла исследование. - Он имеет анатомически правильные регионы, которые развиваются в нужном месте и в нужное время».
Как обнаружили Зерницка-Гетц и ее коллеги, в процессе развития структуры искусственного эмбриона разные типы стволовых клеток проявляют удивительную степень взаимодействия, в каком-то смысле они подсказывают друг другу, в каком месте эмбриона им надлежит оказаться. Трофобластные и эмбриобластные стволовые клетки, использованные в этом эксперименте, были генетически модифицированы: в них были внедрены гены флуоресцентных белков разного цвета, что позволяло следить за их положением.
Сначала эмбриональные стволовые клетки сконцентрировались на одной стороне матрикса, а трофобластные - на противоположной. Затем внутри каждого скопления клеток возникла внутренняя полость. На следующей стадии обе внутренних полости слились, образовав протоамниотическую полость, в которой должно проходить дальнейшее развитие эмбриона. Все эти стадии соответствуют естественному развитию мышиного зародыша. На последнем этапе эксперимента на соответствующем месте внутри искусственного зародыша даже образовались клетки, сходные с примордиальными половыми клетками эмбриона, из которых в дальнейшем должны получиться яйцеклетки или сперматозоиды.
Схема развития зародыша мыши после оплодотворения и развития искусственного зародыша из стволовых клеток и внеклеточного матрикса
Хотя этот искусственный эмбрион очень похож на настоящий, пока он не способен развиваться дальше в здоровый плод. В нынешнем эксперименте развитие эмбриональной структуры продолжалось четыре дня, так как далее искусственному эмбриону не от куда было получать кислород и питательные вещества. Чтобы продолжить его развитие в последующих экспериментах понадобится третья форма стволовых клеток, которые должны образовать особый орган - желточный мешок, обеспечивающий питание эмбриона. При естественном развитии стволовые клетки желточного мешка возникают из клеток эмбриобласта. Также будет необходимо добиться правильного развития плаценты.
Роль легких в кроветворении
Коллектив под руководством профессора Калифорнийского университета в Сан-Франциско Марка Луни доказал, что в легких образуется более половины тромбоцитов - элементов крови, необходимых для ее нормального свертывания. До этого единственным местом образования тромбоцитов считался костный мозг. Также ученые обнаружили в легких неизвестную ранее группу стволовых клеток крови, способную восстановить ее производство, когда с этим не справляются клетки костного мозга. Наблюдения, которые привели к такому открытию, были проведены на лабораторных мышах, но профессор Луни считает, что данная функция легких имеется и у людей.
Открытие стало возможным благодаря применению техники, получившей название «двухфотонная прижизненная визуализация». Ее разработали Марк Луни и Мэтью Краммел (Matthew F. Krummel). Технология позволяет отслеживать отдельные клетки внутри капилляров у живых мышей. У генетически модифицированных мышей тромбоциты обладали способностью к флуоресценции ярко-зеленым цветом. Поэтому ученые смогли отслеживать в живых мышах скопления мегакариоцитов - особых клеток, служащих для производства тромбоцитов. Группа этих клеток в легких мыши выпускает в кровеносную систему более десяти миллионов тромбоцитов в час. Клетки-предшественники мегакариоцитов и другие стволовые клетки крови обнаружились внутри легких вне кровеносных сосудов. Их насчитывается до миллиона в одном мышином легком.
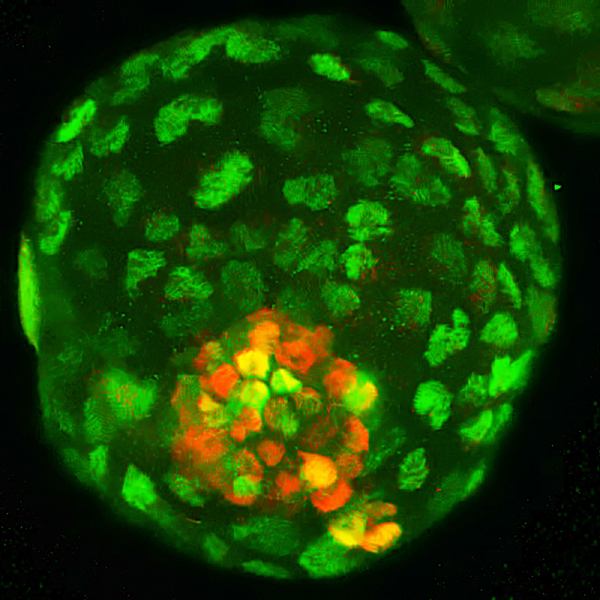
Мегакариоциты в легких
Марк Луни и его коллеги провели несколько операций по трансплантации легких. Сначала они пересадили легкие от обычных мышей мышам с флуоресцентными мегакариоцитами и обнаружили, что флуоресцентные мегакариоциты у мышей-реципиентов вскоре начали появляться в сосудистой системе легкого. Это означало, что тромбоцитарные мегакариоциты в легком происходят из костного мозга. В другом эксперименте были пересажены легкие с флуоресцентными клетками-предшественниками мегакариоцитов мутантным мышам с низким уровнем тромбоцитов. После операции наблюдался большой всплеск флуоресцентных тромбоцитов, которые быстро восстановили нормальный уровень содержания тромбоцитов в организме. Эффект сохранялся в течение нескольких месяцев наблюдения - намного дольше, чем продолжительность жизни отдельных мегакариоцитов или тромбоцитов. Значит клетки-предшественники мегакариоцитов в трансплантированных легких активизировались под влиянием низкого уровня тромбоцитов у мышей-реципиентов и создали здоровые новые мегакариоцитные клетки для восстановления надлежащего количества тромбоцитов.
Наконец, исследователи трансплантировали здоровые легкие, в которых все клетки были флуоресцентно маркированными, мышам, в костном мозге которых отсутствовали нормальные стволовые клетки крови. После этого флуоресцентные клетки из пересаженных легких вскоре переместились в поврежденный костный мозг и способствовали производству не только тромбоцитов, но и самых разнообразных клеток крови, включая иммунные клетки, такие как нейтрофилы, В-клетки и Т-клетки.
Эти эксперименты позволяют предположить, что в легких содержится большое количество стволовых клеток, способных восстанавливать поврежденный костный мозг и вновь начинать выработку многих компонентов крови. Итоги работы важны для многих людей, страдающих тромбоцитопенией - снижением числа тромбоцитов, вызывающим проблемы со свертываемостью крови.
Стволовые клетки крови впервые получены в лаборатории
После двадцати лет попыток ученым впервые удалось превратить соматические клетки в клетки-предшественники клеток крови. Такие клетки, называемые также гемопоэтическими стволовыми клетками, способны дать начало любым другим клеткам крови. Если данный метод удастся применить в клинических условиях, он даст шанс на излечения больных с лейкемией и другими заболеваниями крови, которым требуется пересадка костного мозга, но не находится подходящих доноров.
Об успехе сообщили сразу две исследовательские группы. Коллектив под руководством известного специалиста по стволовым клеткам Джорджа Дейли (George Daley) в Бостонской детской больнице Массачусетса создала клетки человека, которые действуют как стволовые клетки крови, хотя они не идентичны тем, что встречаются в природе. Вторая группа, которую возглавляет профессор медицинской генетики Шахин Рафии (Shahin Rafii) из Медицинского колледжа Уэйлл-Корнелл в Нью-Йорке, превратила зрелые соматические клетки мышей в полноценные стволовые клетки крови. Обе работы опубликованы недавно в журнале Nature.
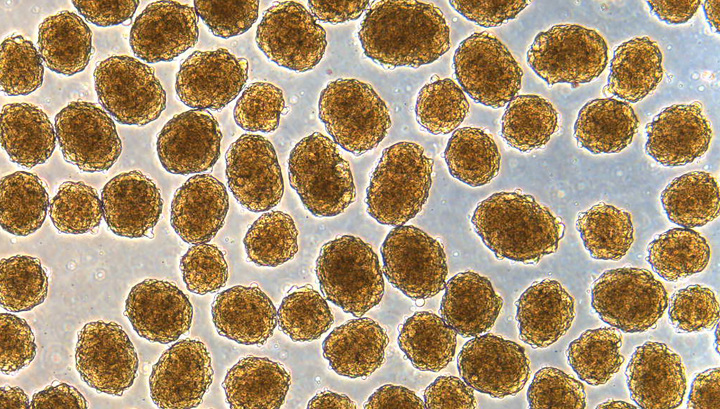
Выращенные в лаборатории стволовые клетки крови под микроскопом
В качестве исходного материала группа Дейли использовала клетки кожи и другие клетки, взятые у взрослых пациентов. Используя стандартный метод, они перепрограммировали эти клетки в индуцированные плюрипотентные стволовые клетки (iPS-клетки), которые способны далее дифференцироваться в другие типы клеток. Однако до последнего времени не удавалось превратить iPS-клетки в гемопоэтические стволовые клетки.
Чтобы добиться этого, понадобилось применить новинку. В геномы iPS-клеток Дейли и его коллеги ввели семь дополнительных генов, кодирующих факторы транскрипции, то есть контролирующих работу других генов. Факторы транскрипции - это белки, способные связываться с определенными участками ДНК и воздействовать на процесс считывания генетической информации при формировании матричной РНК, понижая или повышая активность конкретного гена.
Модифицированные таким образом человеческие клетки были введены для дальнейшего развития в организм лабораторных мышей. В итоге в эксперименте Джорджа Дейли и его коллег iPS-клетки через 12 дней дифференцировались в клетки-предшественники, способные давать начало самым разным клеткам человеческой крови, включая иммунные клетки, например, Т- и В-лимфоциты. По словам ученых, полученные клетки-предшественники очень близки естественным гемопоэтическим стволовым клеткам.
В работе Шахина Рафии и его коллег iPS-клетки не использовались. Они взяли у взрослых мышей клетки эндотелия - слоя толщиной в одну клетку, выстилающего внутреннюю поверхность кровеносных сосудов. В геномы взятых клеток были введены гены четырех дополнительных факторов транскипции. Затем полученные клетки выращивались в лаборатории в чашках Петри, среда которых имитировала условия внутри кровеносных сосудов человека. Там эти клетки превращались в стволовые клетки крови и размножались.
Для проверки команда Рафии ввела полученные клетки мышам, у которых предварительно были убиты радиацией собственные стволовые клетки крови, находящиеся в костном мозге. После введения новых клеток мыши выздоравливали, у них восстанавливался полный набор необходимых клеток крови и они благополучно жили полноценной жизнью, прожив в лаборатории более полутора лет.
Шахин Рафии сравнивает свой метод с прямым авиарейсом, тогда как метод, использующий стадию iPS-клеток, с самолетом, который сначала облетает вокруг Луны, а лишь потом следует к месту назначения. К тому же в методе Рафии требуется введение в геном клеток меньшего числа дополнительных генов, а каждый новый такой ген осложняет работу, ведь часть клеток оказывается неспособной его принять.
Однако Дейли и его коллеги считают, что их способ можно сделать более эффективным, к тому же, по их мнению, после доработки он с меньшей вероятностью будет вызывать опухоли и другие аномалии в модифицированных клетках. Путь к этому они видят во временной регуляции экспрессии генов в iPS-клетках, которая должна прийти на смену постоянному добавлению в их геном генов факторов транскрипции. Также они отмечают, что iPS-клетки могут быть получены из кожи и других тканей, к которым легко получить доступ, тогда как метод Рафии использует клетки с внутренней части кровеносных сосудов, которые труднее добыть и сохранить в лаборатории.
Пока рано говорить, чей метод в итоге окажется более успешным. Но обе публикации значительно повысили оптимизм ученых относительно создания стволовых клеток крови. До последнего времени длительные безуспешные попытки в этом направлении заставляли многих думать, что это сделать вовсе невозможно.
Первые испытания персонализированных вакцин против меланомы
Получены первые сообщения об успехе клинических испытаний вакцин, предназначенных для борьбы с меланомой. Вакцины, адаптированные к индивидуальным мутациям пациента, обучают его иммунную систему борьбе с опухолевыми клетками. Одно из испытаний было проведено в США под руководством Кэтрин У, Патрика Отта и Чжутин Ху, второе провели исследователи из Германии и Австрии. Принципы работы вакцин против меланомы аналогичны вакцинам, направленным против инфекционных заболеваний. Уникальные компоненты опасного для организма объекта - в данном случае опухолевых клеток - смешиваются с агентами, которые стимулируют иммунный ответ. Но данные противораковые вакцины адаптируются к каждому пациенту индивидуально и вводятся уже после появления у него меланомы, а не с профилактической целью.
В обоих случаях исследователи сначала секвенировали гены опухолевых клеток, связанные с кодированием определенных белков, которые наиболее вероятно вызовут иммунный ответ. В американском исследовании приняли участие шесть пациентов, перенесших операцию по удалению опухоли. Обычно рецидивы после таких операций возникают у половины больных. Для каждого участника была подготовлена вакцина, содержащая до 20 фрагментов белков, соответствующих мутациям в их опухолях. Среди шестерых пациентов, получивших персонализированную вакцину, у четверых в течение года не было отмечено новых признаков меланомы. Те же двое, у которых меланома появилась вновь, полностью от нее избавились после приема препарата, который активизировал иммунную систему, блокируя белок PD-1.
Германско-австрийская группа, которую возглавлял профессор Угур Шахин (U) из Университета имени Гуттенберга в Майнце, включила в испытание 13 пациентов с меланомой. Вакцина, которую они использовали, включала РНК, кодирующую до десяти мутированных белков у каждого пациента. У восьми пациентов, не имевших видимых опухолей во время вакцинации, так и не появились новые опухоли в течение 23 месяцев. У оставшихся пятерых в момент вакцинации были опухоли. После введения вакцины у двоих из них размеры опухолей уменьшились, но у одного позднее возобновились. Однако после применения ингибитора PD-1 у него наступила полная ремиссия.
Хотя в обоих случаях число участников испытаний было очень невелико и отсутствовала контрольная группа, первые результаты можно считать обнадеживающими. Специалист по онкологической иммунологии из Университета Вашингтона в Сент-Луисе Роберт Шрайбер (Robert Schreiber) отмечает, что масштабные испытания уже идут и что исследователи считают особенно перспективным сочетание вакцин с ингибиторами PD-1. Но есть целый ряд нерешенных проблем. На генетическое исследование и подготовку персонализированной вакцины у обеих групп ушло около трех месяцев. Это слишком долгий срок для лечения многих видов рака. Правда, обе группы утверждают, что в состоянии ускорить процесс. По оценке Кэтрин У, время подготовки вакцины можно сократить до шести недель.
.Аналог Ноткоин - TapSwap Получай Бесплатные Монеты
Подробнее читайте на polit.ru
| Источник: polit.ru | Рейтинг новостей: 204 |