2016-8-26 11:52 |
Исследователи из Московского физико-технического института, Института биомедицинской химии, Института энергетических проблем химической физики РАН и ФНКЦ физико-химической медицины представили алгоритм для обнаружения в клетках мутантных белков по данным масс-спектрометрического анализа, сообщается в пресс-релизе МФТИ.
Точечные замены аминокислотных остатков в белках вследствие мутации ДНК могут приводить к изменению функций белков в клетках. Считается, что некоторые из таких изменений становятся ключевыми для последующего развития рака. Изучение мутантных белков поможет найти «слабые места» опухолевых клеток и разработать в дальнейшем более эффективные лекарства.
Исследователи применили технологии «больших данных» в области протеомики - науки, изучающей совокупность белков клетки или целого организма. Основной метод протеомного анализа - масс-спектрометрия, позволяющая точно “взвесить” молекулы белков, пептидов и их фрагментов в образце. В результате получается набор масс-спектров, по виду которых ученому предстоит определить, к каким белкам они относятся. Сделать это исключительно по виду масс-спектра пока невозможно. Поэтому для анализа требуется база данных белковых последовательностей, которые могут встретиться в анализируемом образце. Вместо того, чтобы расшифровывать неизвестную последовательность с нуля, программа просто сверяет экспериментальные данные со “словарем” белковых последовательностей.
Однако этот подход не вполне подходит для поиска белков, аминокислотная последовательность которых отличается от того, что прописано в “эталонном” геноме. Идентифицировать белки с мутациями в раковых клетках невозможно, если база данных не содержит такую форму белка. Тогда в работу вступает протеогеномика - область на стыке протеомики и геномики. Вместо универсальной белковой базы данных в протеогеномных исследованиях используются базы, уникальные для исследуемых клеток, включающие информацию о возможных прочтениях генома и его “перевод” в аминокислотную последовательность.
В новой работе российские ученые разработали алгоритм поиска мутантных белков, позволяющий сравнивать масс-спектрометрические результаты разных исследовательских групп и выделять мутации, связанные с раком. Эффективность подхода исследователи продемонстрировали на клеточной линии HEK-293, полученной из почки человеческого эмбриона. HEK-293 несет множество мутаций и служит отличной моделью для отработки протеогеномного подхода к исследованию рака.
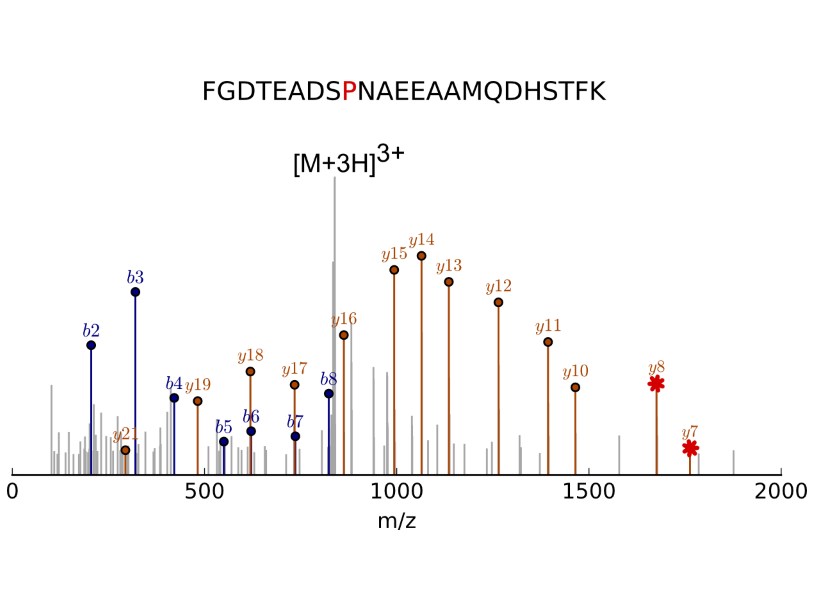
Масс-спектр фрагментов пептида [от белка DMXL2], содержащего точечную мутацию. Красным цветом отмечена позиция мутации (замена аминокислотного остатка S на P) и пики в масс-спектре фрагментации, подтверждающие присутствие данной замены. Илл. : МФТИ
Помимо собственных экспериментальных данных, в своей работе ученые использовали масс-спектры из двух работ, посвященных исследованию протеома клеток HEK-293. Для протеогеномного анализа ученые подготовили расширенную базу данных на основе результатов секвенирования экзома клеточной линии HEK-293. Экзом - это совокупность экзонов (участков генов, которые кодируют последовательность аминокислот в белке). Секвенирование экзома позволяет сконцентрировать внимание исследователей только на белок-кодирующих генах, а также определить расположение экзонов в них. Увеличенная база стала больше на 1336 вариантов последовательностей. Таким образом в белковый “словарь” добавились последовательности, которые отличаются от исходных на одну или несколько “букв” - аминокислот. Без этой поправки поисковая программа не смогла бы обнаружить такие немного “неправильные” белки. Поскольку в каждой клетке постоянно происходят мутации, а в раковых - особенно часто, обнаружение белков, отличающихся от “эталонных”, поможет понять, чем опухолевая клетка отличается от нормальной.
По данным масс-спектрометрического анализа из двух ранних исследований и экспериментально полученным в текущей работе масс-спектрам ученые определили, какие пептиды - короткие белки или фрагменты белков - содержатся в клетках и к каким белкам они относятся. Применяя новый подход к протеогеномному анализу с использованием расширенной базы данных, ученые обнаружили в клетках HEK-293 113 уникальных вариантных последовательностей пептидов, относившихся к экзонным областям 103 генов. Это существенно больше, чем в работе, ранее выполненной группой Стивена Гиги, где не использовалась дополненная геномными вариантами белковая база данных, а поиск аминокислотных замен производился другим методом.
Для некоторых из обнаруженных мутаций ранее была показана связь с различными типами рака. Возможно, наличие этих вариантов способствует лучшему выживанию и размножению клеток. В частности, один из обнаруженных геномных вариантов относится к белку p53, подавляющему злокачественное преобразование клетки.
«Наш подход может в дальнейшем использоваться для поиска ассоциированных с раком мутаций на основе протеомных данных. Это, в свою очередь, поможет в изучении белкового состава опухолей и разработке препаратов, “нацеленных” на мутантные белки, производимые в опухолевых клетках», - отмечает Михаил Горшков, один из руководителей проекта, заведующий лабораторией физико-химических методов исследования структуры веществ ИНЭПХФ РАН, сотрудник Кафедры химической физики МФТИ.
Результаты исследования опубликованы в журнале Proteomics.
.Аналог Ноткоин - TapSwap Получай Бесплатные Монеты
Подробнее читайте на polit.ru
| Источник: polit.ru | Рейтинг новостей: 214 |